Корреляция генотипа и фенотипа при врожденном амаврозе Лебера
Leber congenital amaurosis: genotype-phenotype correlations
A cohort of 58 Dutch and Belgian LCA patients was genotyped using a newly
developed LCA APEX microarray (chapter 3), and mutations were detected in 33%
of the cases. In a study performed by Hanein and co-workers, a larger number of
LCA patients was screened using a more comprehensive but much more labor
intensive approach (linkage analysis, dHPLC, direct sequencing), which revealed
mutations in 44% of the cases21. Assuming that 44% is the maximum percentage of
patients in which variants can be found today, we can conclude that the LCA APEX
microarray detected 75% of the expected mutations.
In our patient cohort, mutations were most frequently identified in the CRB1 gene
(16%) and all carried either the p.C984Y or the p.K801X mutation. The second most
frequently involved gene was GUCY2D. The GUCY2D p.R768W mutation appears
to be a founder mutation in the Northwest of Europe, because of it unusually high
frequency. In previous studies, mutations in CRB1 were found to underlie 10-13%
of the LCA cases21-23. Recently, den Hollander and co-workers analysed 24 French-
Canadian LCA patients and found only one mutated allele22. On the other hand, a
Spanish study revealed CRB1 to be the causative gene in ~2/3 of all LCA patients
with proven mutations, suggesting that the CRB1 gene is a major LCA causing
gene in Western Europe (Vallespin E, personal communication, 2005). The latter two
studies illustrate extensive ethnic and geographical differences in the occurrence of
CRB1 mutations.
Specific genotype-phenotype correlations were made for three
LCA genes. Patients with AIPL1 mutations showed very poor visual acuity and
hypermetropia with essentially normal appearing fundi on initial examination. After
the age of eight years, extensive pigmentations became present with a striking
bulls’ eye maculopathy. All patients with CRB1 mutations showed some or all signs
of the RP12 phenotype, being relatively normal appearing optic discs and vessels,
a distinct maculopathy, a nummular type of pigmentations, yellow-white subretinal
dots, PPRPE, Coats-like exudative vasculopathy and vascular sheathing. LCA
patients with GUCY2D mutations showed an extremely severe phenotype of no light
perception and relatively normal appearing fundi until at least the age of 30 years.
In a relatively large LCA cohort, Hanein and co-workers suggested that symptoms
such as night blindness and photophobia may be distinctive symptoms that point
to the gene involved23. Our study did not confirm this correlation. Night blindness
or photophobia as gene-specific characteristics may be applicable to a group of
patients with a certain genotype; however they cannot be used as good predictors
in the individual patient. Unfortunately, although we presented specific genotypephenotype
correlations, it may still be very difficult to predict the gene involved in a
single LCA patient after one clinical examination. Our study, although small in size,
did indicate that there are trends in the correlation between LCA gene defects and
the resulting phenotype. This correlation likely becomes more complete in the near
future, as more detailed structural (optical coherence tomography) and functional
(autofluorescence) imaging techniques are now available. These results, together
with a longer follow up of the LCA patients, will add new phenotypic aspects to the
genotype-phenotype correlations. In the future, more genotype-phenotype studies
need to be performed in order to establish better indicators for which specific gene
is involved.
The gene encoding the retinal pigment epithelium-specific 65-kDa
protein (RPE65) was studied. RPE65 is the only gene studied in this thesis that
is expressed exclusively in the RPE. The most detailed phenotypic description of
patients suffering from autosomal recessive early-onset retinal dystrophy caused
by RPE65 mutations consistently showed preserved vision in childhood with
a useful visual function beyond the second decade of life27, which later declined.
Interfamilial variations in the RPE65-associated phenotype were found in these
studies, which were attributed to the effects of different types of mutations (different
amount of residual protein function). Authors reported on the existence of intrafamilial variations in patients with
the same homozygous mutation in RPE65. In a large, previously isolated Dutch
population a p.Y368H RPE65 mutation was detected homozygously in 13 out of 14
patients from 10 related families. The patients all showed signs of night blindness,
nystagmus, severe visual impairment with relatively well-preserved visual fields and a
non-detectable ERG in early childhood. On fundoscopy, the older patients showed
optic disc pallor, attenuated vessels and a hypopigmented periphery. Surprisingly, a
wide range of visual acuity (VA) and natural history of visual function was observed.
Approximately one third of the patients showed mild improvement, one third showed
deterioration and one third showed a stable VA over a mean 9-year follow-up
period. Clearly, other genetic and/or environmental factors must be involved in the
heterogeneous phenotype in this pedigree.
Further studies of this population revealed the founder mutation heterozygously in
1/28 of healthy controls rendering a risk of 1 in 56 for the child of a patient with a
partner from this population to develop the retinal dystrophy due to homozygous
RPE65 mutations. Likewise, heterozygous carriers with partners from this population
have a risk of 1 in 112 that any of their children will develop the disease. Recently,
additional patients from this population with either arRP or early-onset RP were
ascertained. Sequence analysis of the RPE65 gene revealed new mutations in this
gene, rendering the relative burden of RPE65 mutations in this population even
higher (S. Yzer, F.P.M. Cremers, unpublished data). Our studies now enable p.Y368H
carrier detection in this population, which theoretically could lead to a significant
decrease of children born with this severe sensory handicap.
It is estimated that autosomal recessive mutations in RPE65 account for 6-16%
of the LCA cases in the world. The identification of patients with RPE65 defects has
become very important since gene therapy studies in RPE65 deficient animal models
proved to restore photoreceptor function both on observational and functional (ERG)
studies30-32. A human clinical trial for single dose subretinal delivery of recombinant
adeno-associated virus carrying wild-type RPE65 cDNA, is not far off.
As illustrated in authors studies, LCA is a remarkably heterogeneous disease, both
clinically and genetically. Genotyping-phenotyping efforts as presented in this
thesis will ultimately lead to a more detailed knowledge facilitating phenotypical
differentiation. Together with new technologies these efforts will facilitate and speed
up the molecular diagnostic process. Unfortunately, the current knowledge on LCA
phenotypes does not provide enough information for accurate gene selection in
individual patients. In order to aid a more profound genotype-phenotype correlation
in the future, more mutations need to be identified, other LCA genes need to be
identified and additional genotype-phenotype correlation studies are needed. Until
then, the most logical and most cost-effective genotyping approach would be to
have a patients’ DNA tested using the APEX LCA microarray.
Carriers of LCA mutations show distinctive functional
abnormalities
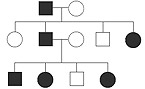
Classical genetic definitions (Mendelian inheritance) teach that recessive mutations
only cause a phenotype in the individual that carries two defective genes. A carrier
with only one copy of the defective allele is thought to be protected by the wild-type
allele and therefore show no obvious disease signs. Obligate heterozygous carriers
of LCA mutations in the AIPL1, GUCY2D, RDH12 and RPGRIP1 genes, however,
were shown to have distinct ERG abnormalities33,34, without any other signs or
symptoms. Carriers of AIPL1 mutations showed abnormal rod amplitudes whereas
carriers of heterozygous GUCY2D pathogenic sequence changes showed cone
abnormalities, as did carriers of RDH12 variants. RPGRIP1 heterozygotes showed
both rod and cone ERG dysfunction34. In contrast to carriers of AIPL1, GUCY2D,
RDH12 and RPGRIP1 pathogenic sequence changes, the heterozygous carriers of
RPE65 mutations show normal ERG function35-37. Apparently, the presence of only
one wild-type LCA allele in most families is not enough to suppress a (sub)clinical
phenotype. Our goal in chapter 5 was to elucidate the possible CRB1 heterozygous
phenotype.
Two different Crb1 animal models were previously studied. In a natural Crb1 mutant
mouse, the predicted Crb1 protein is truncated lacking the transmembrane and
intracellular domain. Histologically, these animals show irregularities at the outer
limiting membrane and loss of the photoreceptors, eventually resulting in multiple
intraretinal pseudorosettes localized to the inferior nasal quadrant of the fundus.
In the second mouse model both Crb1 alleles were inactivated by deleting a 2.9 kb
segments of genomic Crb1 sequence containing the upstream promoter region,
exon 1 (encoding the start of the protein) and part of intron 1. This model shows
giant half rosettes in the inferior temporal quadrant but no loss of overall retinal
function on ERG 37.
In chapter 5 obligatory carriers of CRB1 mutations were tested for gene specific
electroretinography abnormalities. Since the retinal dystrophies in the mouse models
were localized we decided to use a specific mfERG (cone) pattern for testing the
heterozygous CRB1 mutation carriers. Five out of 7 obligatory carriers demonstrated
specific infero-nasal mosaic retinal dysfunction. The other two carriers showed no
abnormalities on mfERG recordings.
The previously mentioned mouse model studies also provided information on
Crb1 to be essential in the maintenance of the adherens junctions between
photoreceptors and Muller glia cells during light exposure. It was therefore proposed
that Crb1 might play a crucial role in the prevention of retinal disorganization and
retinal dystrophy. Light may consequently influence the development of retinal
disease in the presence of CRB1 mutations37. Difference in light exposure may for
that reason also play a role in the mfERG recording differences in the tested CRB1
mutation carriers.
Interestingly, McKay and co-workers recently reported on a family with autosomal
dominant inherited pigmented paravenous chorioretinal atrophy due to a p.V62M
mutation in the CRB1 gene38. This is of special interest because the chorioretinal
atrophy was subtle in the early disease stage and limited to the inferior quadrant.
Disease expression was variable and males where more likely to exhibit a severe
phenotype compared to females which remained virtually asymptomatic.
Although the cohort of heterozygous CRB1 carriers studied is rather small, the
majority of our tested individuals display an abnormal phenotype. Moreover, this
phenotype is different from previously described heterozygous carrier phenotypes
in other tested LCA genes. In the future, mfERG testing of additional heterozygous
carriers may specify the ocular carrier phenotype and may also provide
characteristic findings in the parents of LCA offspring and add important information
on the gene involved in their offspring. MfERG testing in parents of LCA children may
therefore be an informative tool in differentiating which gene is involved, potentially
facilitating the molecular diagnostic process.
This thesis has supplied information on the usefulness and efficiency of newly
developed microarrays that facilitate molecular diagnostics in a portion of autosomal
recessive retinopathies. Furthermore, important clinical data on ABCA4 associated
arCRD and arRP as for AIPL1, CRB1 and GUCY2D associated LCA and RPE65
associated early-onset rod-cone dystrophy were provided.
In conclusion, molecular and clinical studies of retinal diseases provide crucial
knowledge for the understanding of normal and abnormal retinal development and
function.
Подробнее в книге Autosomal recessive retinal dystrophies: genotypes & phenotypes (скачать, pdf 11 Мб) авторов Kortmann, Suzanne IJzer, Frans Cremers (Нидерланды, Radboud Nijmegen Университет).
|